The term sickle cell disease (SCD) describes a group of inherited red blood cell disorders. People with SCD have abnormal hemoglobin, called hemoglobin S or sickle hemoglobin, in their red blood cells.
Hemoglobin is a protein in red blood cells that carries oxygen throughout the body.
“Inherited” means that the disease is passed by genes from parents to their children. SCD is not contagious. A person cannot catch it, like a cold or infection, from someone else.
People who have SCD inherit two abnormal hemoglobin genes, one from each parent. In all forms of SCD, at least one of the two abnormal genes causes a person’s body to make hemoglobin S. When a person has two hemoglobin S genes, Hemoglobin SS, the disease is called sickle cell anemia. This is the most common and often most severe kind of SCD.
Hemoglobin SC disease and hemoglobin Sβ thalassemia (thal-uh-SEE-me-uh) are two other common forms of SCD.
Some Forms of Sickle Cell Disease
- Hemoglobin SS
- Hemoglobin SC
- Hemoglobin Sβ0 thalassemia
- Hemoglobin Sβ+ thalassemia
- Hemoglobin SD
- Hemoglobin SE
Overview
Cells in tissues need a steady supply of oxygen to work well. Normally, hemoglobin in red blood cells takes up oxygen in the lungs and carries it to all the tissues of the body.
Red blood cells that contain normal hemoglobin are disc shaped (like a doughnut without a hole). This shape allows the cells to be flexible so that they can move through large and small blood vessels to deliver oxygen.
Sickle hemoglobin is not like normal hemoglobin. It can form stiff rods within the red cell, changing it into a crescent, or sickle shape.
Sickle-shaped cells are not flexible and can stick to vessel walls, causing a blockage that slows or stops the flow of blood. When this happens, oxygen can’t reach nearby tissues.
Normal Red Cells and Sickle Red Cells
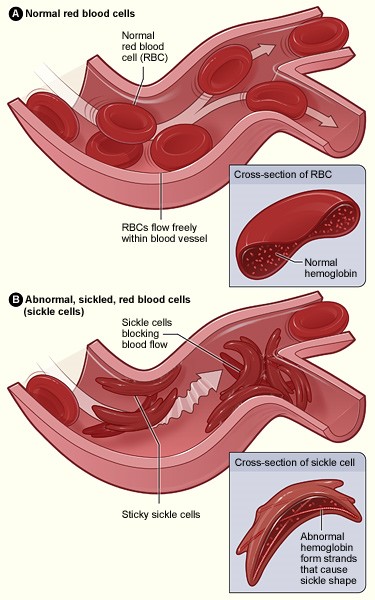
The lack of tissue oxygen can cause attacks of sudden, severe pain, called pain crises. These pain attacks can occur without warning, and a person often needs to go to the hospital for effective treatment.
Most children with SCD are pain free between painful crises, but adolescents and adults may also suffer with chronic ongoing pain.
The red cell sickling and poor oxygen delivery can also cause organ damage. Over a lifetime, SCD can harm a person’s spleen, brain, eyes, lungs, liver, heart, kidneys, penis, joints, bones, or skin.
Sickle cells can’t change shape easily, so they tend to burst apart or hemolyze. Normal red blood cells live about 90 to 120 days, but sickle cells last only 10 to 20 days.
The body is always making new red blood cells to replace the old cells; however, in SCD the body may have trouble keeping up with how fast the cells are being destroyed. Because of this, the number of red blood cells is usually lower than normal. This condition, called anemia, can make a person have less energy.
Outlook
Sickle cell disease is a life-long illness. The severity of the disease varies widely from person to person.
In high-income countries like the United States, the life expectancy of a person with SCD is now about 40–60 years. In 1973, the average lifespan of a person with SCD in the United States was only 14 years. Advances in the diagnosis and care of SCD have made this improvement possible.
At the present time, hematopoietic stem cell transplantation (HSCT) is the only cure for SCD. Unfortunately, most people with SCD are either too old for a transplant or don’t have a relative who is a good enough genetic match for them to act as a donor. A well-matched donor is needed to have the best chance for a successful transplant.
There are effective treatments that can reduce symptoms and prolong life. Early diagnosis and regular medical care to prevent complications also contribute to improved well-being.
Other Names for Sickle Cell Anemia
- HbS disease
- Hemoglobin S disease
- Hemoglobin SS disease
- Sickle cell disease (a broad term that includes sickle cell anemia)
- Sickle cell disorders (a broad group of conditions that includes sickle cell anemia)
- Sickling disorder due to hemoglobin S
What Causes Sickle Cell Disease?
Abnormal hemoglobin, called hemoglobin S, causes sickle cell disease (SCD).
The problem in hemoglobin S is caused by a small defect in the gene that directs the production of the beta globin part of hemoglobin. This small defect in the beta globin gene causes a problem in the beta globin part of hemoglobin, changing the way that hemoglobin works. (See Overview.)
How Is Sickle Cell Disease Inherited?
When the hemoglobin S gene is inherited from only one parent and a normal hemoglobin gene is inherited from the other, a person will have sickle cell trait. People with sickle cell trait are generally healthy.
Only rarely do people with sickle cell trait have complications similar to those seen in people with SCD. But people with sickle cell trait are carriers of a defective hemoglobin S gene. So, they can pass it on when they have a child.
If the child’s other parent also has sickle cell trait or another abnormal hemoglobin gene (like thalassemia, hemoglobin C, hemoglobin D, hemoglobin E), that child has a chance of having SCD.
Example of an Inheritance Pattern
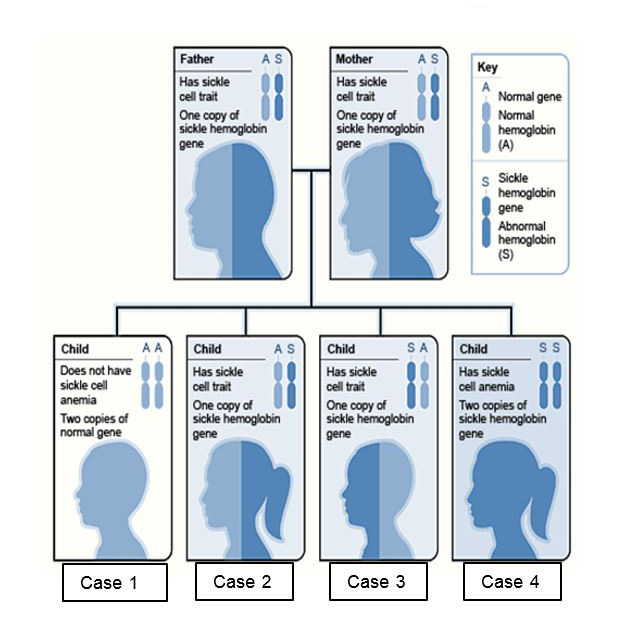
In the image above, each parent has one hemoglobin A gene and one hemoglobin S gene, and each of their children has:
- A 25 percent chance of inheriting two normal genes: In this case the child does not have sickle cell trait or disease. (Case 1)
- A 50 percent chance of inheriting one hemoglobin A gene and one hemoglobin S gene: This child has sickle cell trait. (Cases 2 and 3)
- A 25 percent chance of inheriting two hemoglobin S genes: This child has sickle cell disease. (Case 4)
It is important to keep in mind that each time this couple has a child, the chances of that child having sickle cell disease remain the same. In other words, if the first-born child has sickle cell disease, there is still a 25 percent chance that the second child will also have the disease. Both boys and girls can inherit sickle cell trait, sickle cell disease, or normal hemoglobin.
If a person wants to know if he or she carries a sickle hemoglobin gene, a doctor can order a blood test to find out.
Who Is at Risk for Sickle Cell Disease?
In the United States, most people with sickle cell disease (SCD) are of African ancestry or identify themselves as black.
- About 1 in 13 African American babies is born with sickle cell trait.
- About 1 in every 365 black children is born with sickle cell disease.
There are also many people with this disease who come from Hispanic, southern European, Middle Eastern, or Asian Indian backgrounds.
Approximately 100,000 Americans have SCD.
What Are the Signs and Symptoms of Sickle Cell Disease?
Early Signs and Symptoms
If a person has sickle cell disease (SCD), it is present at birth. But most infants do not have any problems from the disease until they are about 5 or 6 months of age. Every state in the United States, the District of Columbia, and the U.S. territories requires that all newborn babies receive screening for SCD. When a child has SCD, parents are notified before the child has symptoms.
Some children with SCD will start to have problems early on, and some later. Early symptoms of SCD may include:
- Painful swelling of the hands and feet, known as dactylitis
- Fatigue or fussiness from anemia
- A yellowish color of the skin, known as jaundice, or whites of the eyes, known as icteris, that occurs when a large number of red cells hemolyze
The signs and symptoms of SCD will vary from person to person and can change over time. Most of the signs and symptoms of SCD are related to complications of the disease.
Major Complications of Sickle Cell Disease
Acute Pain (Sickle Cell or Vaso-occlusive) Crisis
Pain episodes (crises) can occur without warning when sickle cells block blood flow and decrease oxygen delivery. People describe this pain as sharp, intense, stabbing, or throbbing. Severe crises can be even more uncomfortable than post-surgical pain or childbirth.
Pain can strike almost anywhere in the body and in more than one spot at a time. But the pain often occurs in the
- Lower back
- Legs
- Arms
- Abdomen
- Chest
A crisis can be brought on by
- Illness
- Temperature changes
- Stress
- Dehydration (not drinking enough)
- Being at high altitudes
But often a person does not know what triggers, or causes, the crisis. (See acute pain management.)
Chronic Pain
Many adolescents and adults with SCD suffer from chronic pain. This kind of pain has been hard for people to describe, but it is usually different from crisis pain or the pain that results from organ damage.
Chronic pain can be severe and can make life difficult. Its cause is not well understood. (See chronic pain management.)
Severe Anemia
People with SCD usually have mild to moderate anemia. At times, however, they can have severe anemia. Severe anemia can be life threatening. Severe anemia in an infant or child with SCD may be caused by:
- Splenic sequestration crisis. The spleen is an organ that is located in the upper left side of the belly. The spleen filters germs in the blood, breaks up blood cells, and makes a kind of white blood cell. A splenic sequestration crisis occurs when red blood cells get stuck in the spleen, making it enlarge quickly. Since the red blood cells are trapped in the spleen, there are fewer cells to circulate in the blood. This causes severe anemia.A big spleen may also cause pain in the left side of the belly. A parent can usually palpate or feel the enlarged spleen in the belly of his or her child.
- Aplastic crisis. This crisis is usually caused by a parvovirus B19 infection, also called fifth disease or slapped cheek syndrome. Parvovirus B19 is a very common infection, but in SCD it can cause the bone marrow to stop producing new red cells for a while, leading to severe anemia.
Splenic sequestration crisis and aplastic crisis most commonly occur in infants and children with SCD. Adults with SCD may also experience episodes of severe anemia, but these usually have other causes.
No matter the cause, severe anemia may lead to symptoms that include:
- Shortness of breath
- Being very tired
- Feeling dizzy
- Having pale skin
Babies and infants with severe anemia may feed poorly and seem very sluggish. (See anemia management.)
Infections
The spleen is important for protection against certain kinds of germs. Sickle cells can damage the spleen and weaken or destroy its function early in life.
People with SCD who have damaged spleens are at risk for serious bacterial infections that can be life-threatening. Some of these bacteria include:
- Pneumococcus
- Hemophilus influenza type B
- Meningococcus
- Salmonella
- Staphylococcus
- Chlamydia
- Mycoplasma pneumoniae
Bacteria can cause:
- Blood infection (septicemia)
- Lung infection (pneumonia)
- Infection of the covering of the brain and spinal cord (meningitis)
- Bone infection (osteomyelitis)
(See how to prevent infections and infection management.)
Acute Chest Syndrome
Sickling in blood vessels of the lungs can deprive a person’s lungs of oxygen. When this happens, areas of lung tissue are damaged and cannot exchange oxygen properly. This condition is known as acute chest syndrome. In acute chest syndrome, at least one segment of the lung is damaged.
This condition is very serious and should be treated right away at a hospital.
Acute chest syndrome often starts a few days after a painful crisis begins. A lung infection may accompany acute chest syndrome.
Symptoms may include:
- Chest pain
- Fever
- Shortness of breath
- Rapid breathing
- Cough
(See acute chest syndrome management.)
Brain Complications
Clinical Stroke
A stroke occurs when blood flow is blocked to a part of the brain. When this happens, brain cells can be damaged or can die. In SCD, a clinical stroke means that a person shows outward signs that something is wrong. The symptoms depend upon what part of the brain is affected. Symptoms of stroke may include:
- Weakness of an arm or leg on one side of the body
- Trouble speaking, walking, or understanding
- Loss of balance
- Severe headache
As many as 24 percent of people with hemoglobin SS and 10 percent of people with hemoglobin SC may suffer a clinical stroke by age 45.
In children, clinical stroke occurs most commonly between the ages of 2 and 9, but recent prevention strategies have lowered the risk. (See Transcranial Doppler (TCD) Ultrasound Screening) and Red Blood Cell Transfusions.)
When people with SCD show symptoms of stroke, their families or friends should call 9-1-1 right away. (See clinical stroke management.)
Silent Stroke and Thinking Problems
Brain imaging and tests of thinking (cognitive studies) have shown that children and adults with hemoglobin SS and hemoglobin Sβ0 thalassemia often have signs of silent brain injury, also called silent stroke. Silent brain injury is damage to the brain without showing outward signs of stroke.
This injury is common. Silent brain injury can lead to learning problems or trouble making decisions or holding down a job. (See Cognitive Screening and silent stroke management.)
Eye Problems
Sickle cell disease can injure blood vessels in the eye.
The most common site of damage is the retina, where blood vessels can overgrow, get blocked, or bleed. The retina is the light-sensitive layer of tissue that lines the inside of the eye and sends visual messages through the optic nerve to the brain.
Detachment of the retina can occur. When the retina detaches, it is lifted or pulled from its normal position. These problems can cause visual impairment or loss. (See Eye Examinations.)
Heart Disease
People with SCD can have problems with blood vessels in the heart and with heart function. The heart can become enlarged. People can also develop pulmonary hypertension.
People with SCD who have received frequent blood transfusions may also have heart damage from iron overload. (See transfusion management.)
Pulmonary Hypertension
In adolescents and adults, injury to blood vessels in the lungs can make it hard for the heart to pump blood through them. This causes the pressure in lung blood vessels to rise. High pressure in these blood vessels is called pulmonary hypertension. Symptoms may include shortness of breath and fatigue.
When this condition is severe, it has been associated with a higher risk of death. (See screening for pulmonary hypertension.)
Kidney Problems
The kidneys are sensitive to the effects of red blood cell sickling.
SCD causes the kidneys to have trouble making the urine as concentrated as it should be. This may lead to a need to urinate often and to have bedwetting or uncontrolled urination during the night (nocturnal enuresis). This often starts in childhood. Other problems may include:
- Blood in the urine
- Decreased kidney function
- Kidney disease
- Protein loss in the urine
Priapism
Males with SCD can have unwanted, sometimes prolonged, painful erections. This condition is called priapism.
Priapism happens when blood flow out of the erect penis is blocked by sickled cells. If it goes on for a long period of time, priapism can cause permanent damage to the penis and lead to impotence.
If priapism lasts for more than 4 hours, emergency medical care should be sought to avoid complications. (See priapism management.)
Gallstones
When red cells hemolyze, they release hemoglobin. Hemoglobin gets broken down into a substance called bilirubin. Bilirubin can form stones that get stuck in the gallbladder. The gallbladder is a small, sac-shaped organ beneath the liver that helps with digestion. Gallstones are a common problem in SCD.
Gallstones may be formed early on but may not produce symptoms for years. When symptoms develop, they may include:
- Right-sided upper belly pain
- Nausea
- Vomiting
If problems continue or recur, a person may need surgery to remove the gallbladder.
Liver Complications
There are a number of ways in which the liver may be injured in SCD.
Sickle cell intrahepatic cholestasis is an uncommon, but severe, form of liver damage that occurs when sickled red cells block blood vessels in the liver. This blockage prevents enough oxygen from reaching liver tissue.
These episodes are usually sudden and may recur. Children often recover, but some adults may have chronic problems that lead to liver failure.
People with SCD who have received frequent blood transfusions may develop liver damage from iron overload.
Leg Ulcers
Sickle cell ulcers are sores that usually start small and then get larger and larger.
The number of ulcers can vary from one to many. Some ulcers will heal quickly, but others may not heal and may last for long periods of time. Some ulcers come back after healing.
People with SCD usually don’t get ulcers until after the age of 10.
Joint Complications
Sickling in the bones of the hip and, less commonly, the shoulder joints, knees, and ankles, can decrease oxygen flow and result in severe damage. This damage is a condition called avascular or aseptic necrosis. This disease is usually found in adolescents and adults.
Symptoms include pain and problems with walking and joint movement. A person may need pain medicines, surgery, or joint replacement if symptoms persist.
Delayed Growth and Puberty
Children with SCD may grow and develop more slowly than their peers because of anemia. They will reach full sexual maturity, but this may be delayed.
Pregnancy
Pregnancies in women with SCD can be risky for both the mother and the baby.
Mothers may have medical complications including:
- Infections
- Blood clots
- High blood pressure
- Increased pain episodes
They are also at higher risk for:
- Miscarriages
- Premature births
- “Small-for-dates babies” or underweight babies
Mental Health
As in other chronic diseases, people with SCD may feel sad and frustrated at times. The limitations that SCD can impose on a person’s daily activities may cause them to feel isolated from others. Sometimes they become depressed.
People with SCD may also have trouble coping with pain and fatigue, as well as with frequent medical visits and hospitalizations. (See living with emotional issues.)
How Is Sickle Cell Disease Diagnosed?
Screening Tests
People who do not know whether they make sickle hemoglobin (hemoglobin S) or another abnormal hemoglobin (such as C, β thalassemia, E) can find out by having their blood tested. This way, they can learn whether they carry a gene (i.e., have the trait) for an abnormal hemoglobin that they could pass on to a child.
When each parent has this information, he or she can be better informed about the chances of having a child with some type of sickle cell disease (SCD), such as hemoglobin SS, SC, Sβ thalassemia, or others.
Newborn Screening
When a child has SCD, it is very important to diagnose it early to better prevent complications.
Every state in the United States, the District of Columbia, and the U.S. territories require that every baby is tested for SCD as part of a newborn screening program.
In newborn screening programs, blood from a heel prick is collected in “spots” on a special paper. The hemoglobin from this blood is then analyzed in special labs.
Newborn screening results are sent to the doctor who ordered the test and to the child’s primary doctor.
If a baby is found to have SCD, health providers from a special follow-up newborn screening group contact the family directly to make sure that the parents know the results. The child is always retested to be sure that the diagnosis is correct.
Newborn screening programs also find out whether the baby has an abnormal hemoglobin trait. If so, parents are informed, and counseling is offered.
Remember that when a child has sickle cell trait or SCD, a future sibling, or the child’s own future child, may be at risk. These possibilities should be discussed with the primary care doctor, a blood specialist called a hematologist, and/or a genetics counselor.
Prenatal Screening
Doctors can also diagnose SCD before a baby is born. This is done using a sample of amniotic fluid, the liquid in the sac surrounding a growing embryo, or tissue taken from the placenta, the organ that attaches the umbilical cord to the mother’s womb.
Testing before birth can be done as early as 8–10 weeks into the pregnancy. This testing looks for the sickle hemoglobin gene rather than the abnormal hemoglobin.